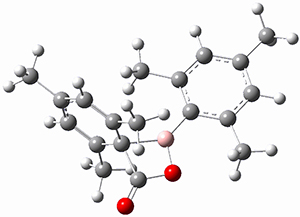
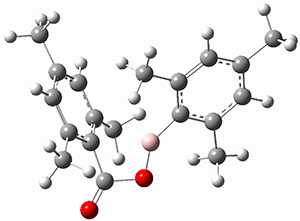
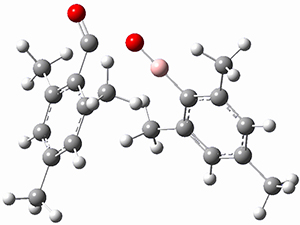
Moritz P. Haag, Alain C. Vaucher, Maël Bosson, Stéphane Redon, Markus Reiher ChemPhysChem 2014, 15, 3301 [arXiv]
Contributed by +Jan Jensen

This work is licensed under a Creative Commons Attribution 4.0 International License.
Contributed by +Jan Jensen
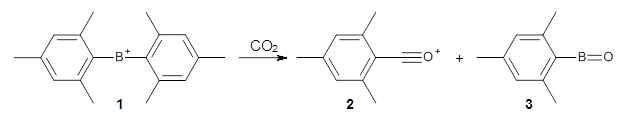
"... it is still a major task to explore reaction mechanisms for molecular systems of even medium size (say, one to a few hundred atoms). ... The trial- and-error approach currently applied (guessing the important structures and refining them with local optimization methods) requires experience, luck, and time."
This paper provides a very innovative solution to this problem by interfacing a haptic device (a Phantom Desktop), the DFTB method, and the SAMSON visualization program to create a tool to interactively find intermediates and transition states.
A haptic device is a force-feedback device that simulates a sense of touch based on computational data - in this case the forces on atoms computed by the DFTB method. This, in essence, allows you to provide physically force the reaction to occur. The premise is that this more intuitive approach to molecular modeling will make it easier for non-experts to quickly and thoroughly explore the reactivity of the system of interest.
In order for this to work convincingly the computational method has to be fast enough to provide real-time feedback on the millisecond timescale and computers and approximate QM methods are now fast enough to provide this. As the user explores the potential energy surface key structures are automatically saved together with their energy and gradient.
Applying this method leads to interesting practical challenges. For example, the Phantom Desktop can only manipulate a single point, so many atomic degrees of freedom must be combined and some degree of automatic energy minimization must be included for most of the atoms. Then there is the "evasive adaption" of the system which naturally favors low energy structures, e.g. by moving atoms out of the way rather than reacting.
I for one would very much like to get my hands on this setup and try this for myself. Also, this really seems like an invaluable teaching tool! It seems like the many years of study required to build up a chemical intuition of reactivity could be replaced by a few weeks with this device! Finally, there is the opportunity of citizen- or crowd sourced-science where non-scientists could help find novel reaction paths, much like fold.it for the protein folding problem.
This work is licensed under a Creative Commons Attribution 4.0 International License.