Malischewski, M.; Seppelt, K., Angew. Chem. Int. Ed. 2017, 56, 368-370
Contributed by Steven Bacharach
Reposted from Computational Organic Chemistry with permission
 '
'
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Reposted from Computational Organic Chemistry with permission
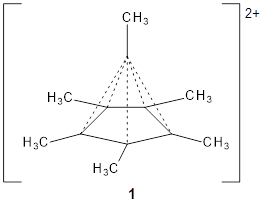
Hypercoordinated carbon has fascinated chemists since the development of the concept of the tetravalent carbon. The advent of superacids has opened up the world of hypercoordinated species and now a crystal structure of a hexacoordinated carbon has been reported for the C6(CH3)62+ species 1.1
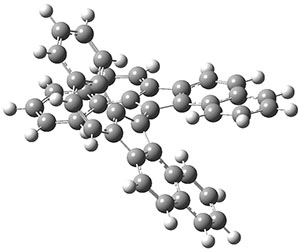
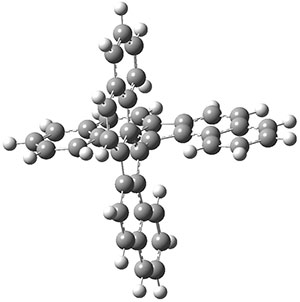
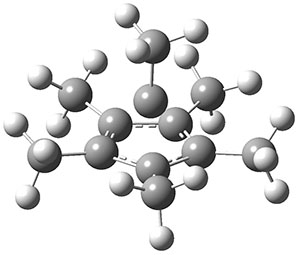
The molecule is prepared by first epoxidation of hexamethyl Dewar benzene, followed by reaction with Magic acid, and crystallized by the addition of HF. The crystal structure shows a pentamethylcyclopentadienyl base capped by a carbon with a methyl group. The x-ray structure is well reproduced by the B3LYP/def2-TZVP structure shown in Figure 1. (While this DFT method predicts a six-member isomer to be slightly lower in energy, MP2 does predict the cage as the lowest energy isomer.)
1
|
Figure 1. B3LYP/def2-TZVP optimized geometry of 1.
The Wiberg bond order for the bond between the capping carbon and each carbon of the five-member base is about 0.54, so the sum of the bond orders to the apical carbon is less than 4. The carbon is therefore not hypervalent, but it appears to truly be hypercoordinate. (A topological electron density analysis (AIM) study would have been interesting here.) NICS analysis indicates the cage formed by the apical carbon and the five-member ring expresses 3-D aromaticity. This can be thought of as coming from the C5(CH3)5+ fragment with its 4 electrons and the CCH3+ fragment with two electrons, providing 4n + 2 = 6 electrons for the aromatic cage.
References
1) Malischewski, M.; Seppelt, K., "Crystal Structure Determination of the Pentagonal-Pyramidal Hexamethylbenzene Dication C6(CH3)62+" Angew. Chem. Int. Ed. 2017, 56, 368-370, DOI: 10.1002/anie.201608795.
'This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.