Contributed by
Steven BacharachReposted from
Computational Organic Chemistry with permission
For the past twelve years, I have avoided posting on any of my own papers, but I will stoop to some shameless promotion to mention my latest paper,
1 since it touches on some themes I have discussed in the past.
Back in 2011, Iwamoto, et al. prepared the complex of C
60 1 surrounded by [10]cycloparaphenylene
2 to make the Saturn-like system
3.
2 Just last year, Yamamoto, et al prepared the Nano-Saturn
5a as the complex of
1 with the macrocycle
4a.
3 The principle idea driving their synthesis was to utilize a ring that is flatter than
2. The structures of
3 and
5b (made with the parent macrocycle
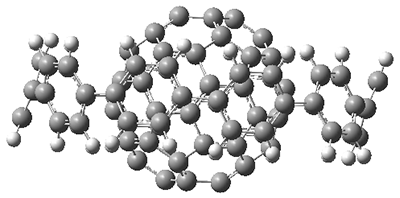
4b) are shown in side view in Figure 1, and clearly seen is the achievement of the flatter ring.
Figure 1. Computed structures of 3, 5, and 7.
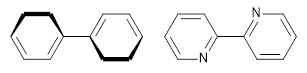
However, the encompassing ring is not flat, with dihedral angles between the anthrenyl groups of 35°. This twisting is due to the steric interactions of the
ortho-ortho’ hydrogens. A few years ago, my undergraduate student David Stück and I suggested that selective substitution of a nitrogen for one of the C-H groups would remove the steric interaction,
4 leading to a planar poly-aryl system, such as making twisted biphenyl into the planar 2-(2-pyridyl)-pyridine (Scheme 1)
Scheme 1.
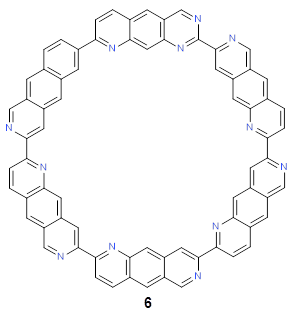
Following this idea leads to four symmetrical nitrogen-substituted analogues of 4b; and I’ll mention just one of them here, 6.
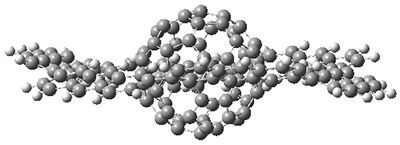
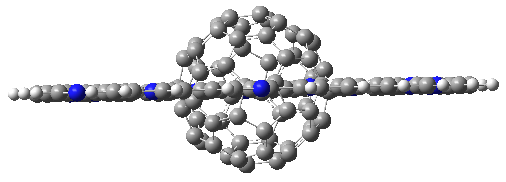
As expected, 6 is perfectly flat. The ring remains flat even when complexed with 1 (as per B3LYP-D3(BJ)/6-31G(d) computations), see the structure of 7 in Figure 1.
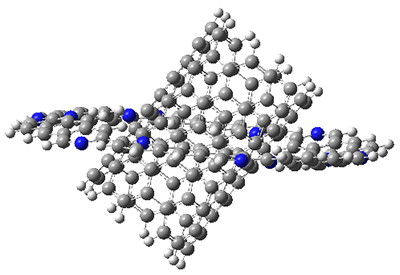
I also examined the complex of the flat macrocycle
6 (and its isomers) with a [5,5]-nanotube,
7. The tube bends over to create better dispersion interaction with the ring, which also become somewhat non-planar to accommodate the tube. Though not mentioned in the paper, I like to refer to
7 as Beyoncene, in tribute to
All the Single Ladies.
Figure 2. Computed structure of 7.
My sister is a graphic designer and she made this terrific image for this work:
References
1. Bachrach, S. M., “Planar rings in nano-Saturns and related complexes.”
Chem. Commun. 2019,
55, 3650-3653, DOI:
10.1039/C9CC01234F.
2. Iwamoto, T.; Watanabe, Y.; Sadahiro, T.; Haino, T.; Yamago, S., “Size-Selective Encapsulation of C
60 by [10]Cycloparaphenylene: Formation of the Shortest Fullerene-Peapod.”
Angew. Chem. Int. Ed. 2011,
50, 8342-8344, DOI:
10.1002/anie.201102302
3. Yamamoto, Y.; Tsurumaki, E.; Wakamatsu, K.; Toyota, S., “Nano-Saturn: Experimental Evidence of Complex Formation of an Anthracene Cyclic Ring with C60.”
Angew. Chem. Int. Ed. 2018 ,
57, 8199-8202, DOI:
10.1002/anie.201804430.
4. Bachrach, S. M.; Stück, D., “DFT Study of Cycloparaphenylenes and Heteroatom-Substituted Nanohoops.”
J. Org. Chem. 2010,
75, 6595-6604, DOI:
10.1021/jo101371m
InChIs
4b: InChI=1S/C84H48/c1-13-61-25-62-15-3-51-33-75(62)43-73(61)31-49(1)50-2-14-63-26-64-16-4-52(34-76(64)44-74(63)32-50)54-6-18-66-28-68-20-8-56(38-80(68)46-78(66)36-54)58-10-22-70-30-72-24-12-60(42-84(72)48-82(70)40-58)59-11-23-71-29-69-21-9-57(39-81(69)47-83(71)41-59)55-7-19-67-27-65-17-5-53(51)35-77(65)45-79(67)37-55/h1-48H
InChIKey=ZYXXLAYETADMDM-UHFFFAOYSA-N
6: InChI=1S/C72H36N12/c1-2-38-14-44-20-45-25-67(73-31-50(45)13-37(1)44)57-9-4-39-15-51-32-74-68(26-46(51)21-61(39)80-57)58-10-5-40-16-52-33-75-69(27-47(52)22-62(40)81-58)59-11-6-41-17-53-34-76-70(28-48(53)23-63(41)82-59)60-12-7-42-18-54-35-77-71(29-49(54)24-64(42)83-60)72-78-36-55-19-43-3-8-56(38)79-65(43)30-66(55)84-72/h1-36H
InChIKey=NSSCKPFBHGOOIJ-UHFFFAOYSA-N

'
This work is licensed under a
Creative Commons Attribution-NoDerivs 3.0 Unported License.