Hashimoto, S.; Nakatsuka, S.; Nakamura, M.; Hatakeyama, T. Angew. Chem. Int. Ed. 2014, 53, 14074-14076
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
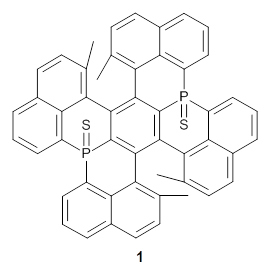
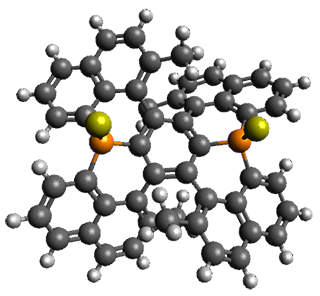
Here’s another cruel and unusual punishment applied to the poor benzene ring. Hashimoto,et al. have created a molecule that is a fused double helicene, where the fusion is about a single phenyl ring.1Compound 1 has two [5]helicenes oriented in opposite directions. This should provide a twist to the central phenyl ring, and the added methyl groups help to expand that twist.
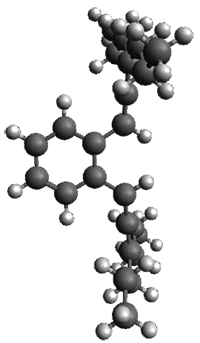
They prepared 1 and its x-ray crystal structure is reported. The compound exhibits C2 symmetry. The twist (defined as the dihedral of four consecutive carbon atoms of the central ring) is 28.17°, nearly the same twist as in [2]paraphenylene.
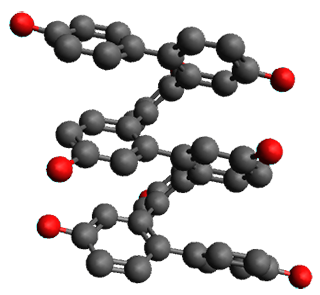
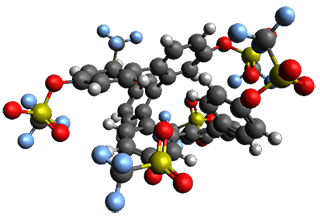
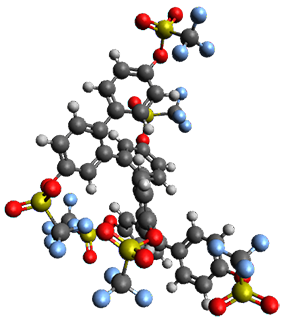
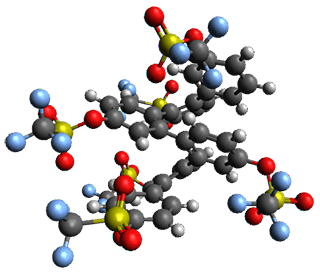
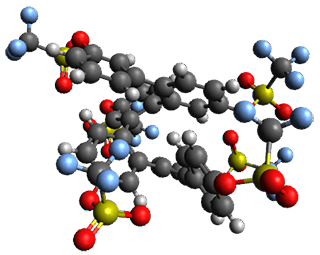
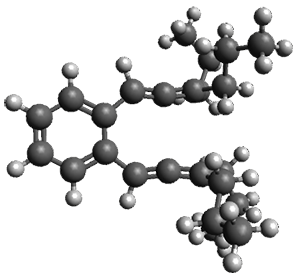
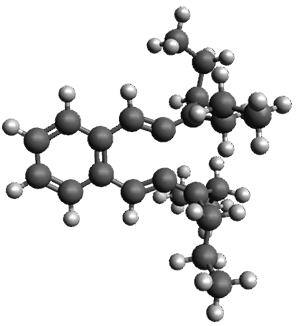
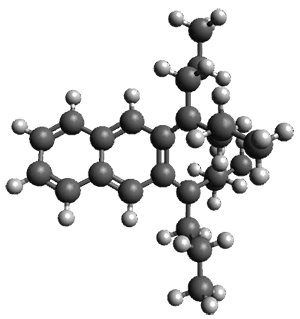
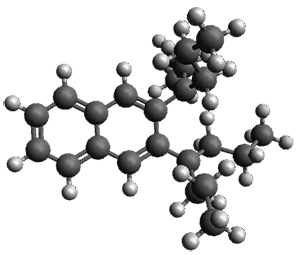
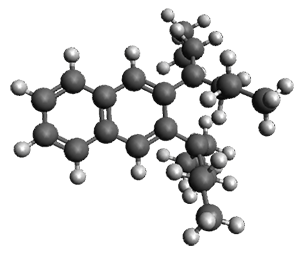
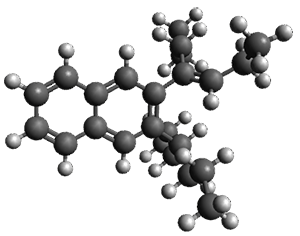
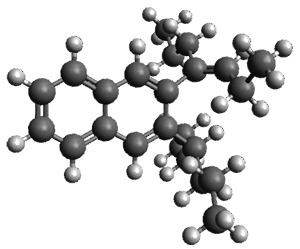
The B3LYP/6-31G(d) structure of 1 is shown in Figure 1. This geometry is very similar to the x-ray structure. The calculated NICS value for the central ring is -4.9 (B3LYP/6-311+G(d,p)/B3LYP/6-31G(d)) and -4.3 (B3LYP/6-311+G(d,p)/x-ray structure). This diminished value from either benzene or C6(PSH2)2(CH3)4 indicates reduced aromaticity of this central ring, presumably due to the distortion away from planarity. Nonetheless, the central ring of 1 is not oxidized when subjected to MCPBA to oxidize to the bis phosphine oxides.
1
|
Figure 1. B3LYP/6-31G(d) optimized structure of 1.
References
(1) Hashimoto, S.; Nakatsuka, S.; Nakamura, M.; Hatakeyama, T. "Construction of a Highly Distorted Benzene Ring in a Double Helicene," Angew. Chem. Int. Ed. 2014, 53, 14074-14076, DOI:10.1002/anie.201408390.
InChIs
1: InChI=1S/C50H32P2S2/c1-25-17-21-29-9-5-13-33-41(29)37(25)45-46-38-26(2)18-22-30-10-7-15-35(42(30)38)52(54)36-16-8-12-32-24-20-28(4)40(44(32)36)48(50(46)52)47-39-27(3)19-23-31-11-6-14-34(43(31)39)51(33,53)49(45)47/h5-24H,1-4H3
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.