Reposted from
Computational Organic Chemistry with permission
I have recently been interested in curved aromatic systems – see my own paper on double helicenes.
1 In this post, I cover four recent papers that discuss non-planar aromatic molecules.
The first paper
2 discusses the warped aromatic
1 built off of the scaffold of depleiadene
3. The crystal structure of
1 shows the molecule to be a saddle with near
C2v symmetry. B3LYP/6-31G computations indicate that the saddle isomer is 10.5 kcal mol
-1 more stable than the twisted isomer, and the barrier between them is 16.0 kcal mol
-1, with a twisted saddle intermediate as well.

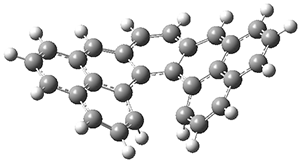
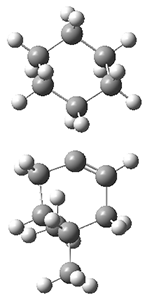
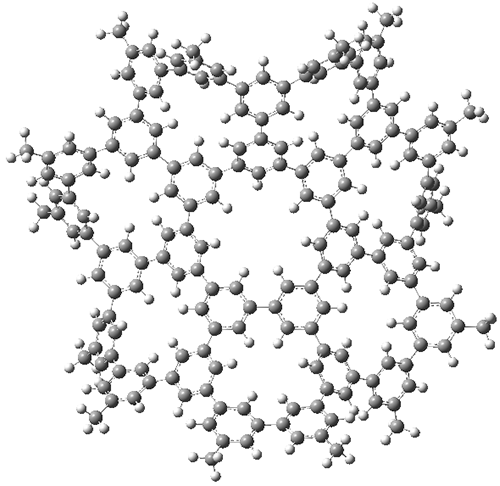
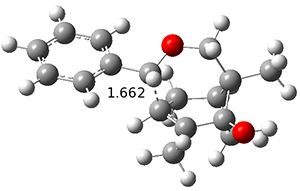
The PES is significantly simpler for the structure lacking the t-butyl groups, 2. The B3LYP/6-31G PES of 2has the saddle as the transition state interconverting mirror images of the twisted saddle isomer, and this barrier is only 1.8 kcal mol-1. Figure 1 displays the twisted saddle and the saddle transition state. Clearly, the t-butyl groups significantly alter the flexibility of this C86 aromatic surface. One should be somewhat concerned about the small basis set employed here, especially lacking polarization functions, and a functional that lacks dispersion correction. However, the computed geometry of 1 is quite similar to that of the x-ray structure.
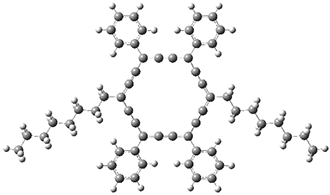
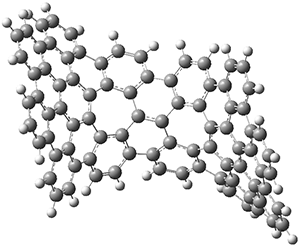 2 2 twisted saddle (ground state)
|
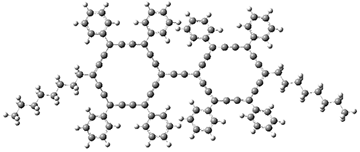
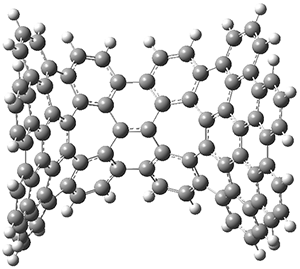 2 2 saddle (transition state)
|
Figure 1. B3LYP/6-31G optimized geometries of the isomer of 2.
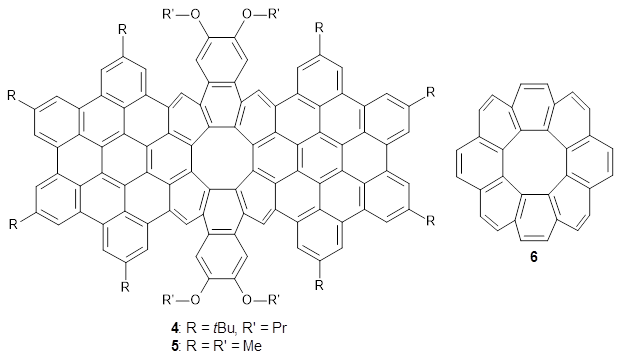
The second paper presents
4, a non-planar aromatic based on [8]circulene
6.
3 (See this
post for a general study of circulenes.) [8]circulene has a tub-shape, but is flexible and can undergo tub-to-tub inversion. The expanded aromatic
4 is found to have a twisted shape in the x-ray crystal structure. A simplified model
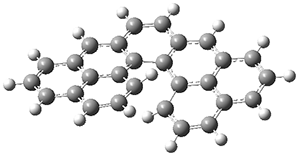
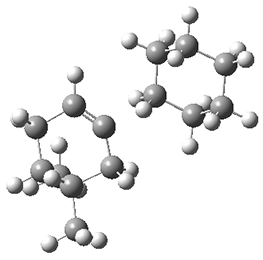
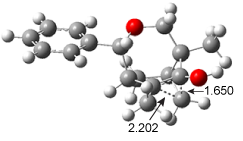
5 was computed at B3LYP/6-31G(d) and the twisted isomer is 4.1 kcal mol
-1 lower in energy than the saddle (tub) isomer (see Figure 2). The barrier for interconversion of the two isomers is only 6.2 kcal mol
-1, indicating a quite labile structure.
 5 twisted 5 twisted0.0
|
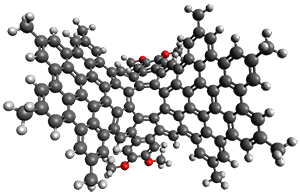 5 TS 5 TS6.2
|
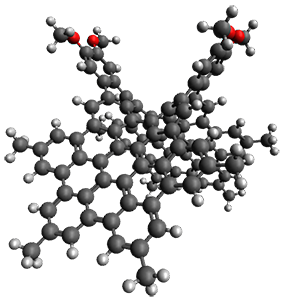 5 saddle 5 saddle4.1
|
Figure 2. B3LYP/6-31G(d) optimized geometries and relative energies (kcal mol-1) of the isomers of 5.
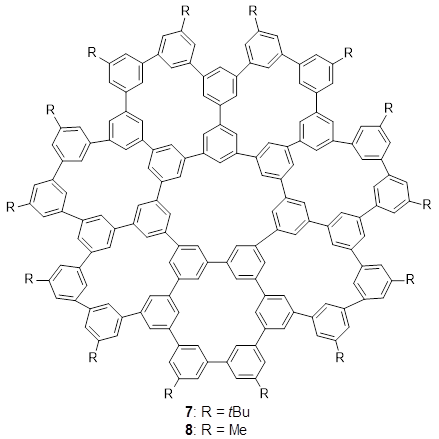
The third paper presents a geodesic molecule based on 1,3,5-trisubstitued phenyl repeat units.
4 The authors prepared
7, and its x-ray structure shows a saddle-shape. The NMR indicate a molecule that undergoes considerable conformational dynamics. To address this, they did some computations on the methyl analogue
8. The
D7h structure is 309 kcal mol
-1 above the local energy minimum structure, which is way too high to be accessed at room temperature. PM6 computations identified a TS only 0.6 kcal mol
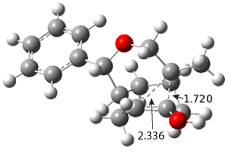
-1above the saddle ground state. (I performed a PM6 optimization starting from the x-ray structure, which is highly disordered, and the structure obtained is shown in Figure 3. Unfortunately, the authors did not report the optimized coordinates of any structure!)
Figure 3. PM6 optimized structure of 8.
The fourth and last paper describes the aza-buckybowl
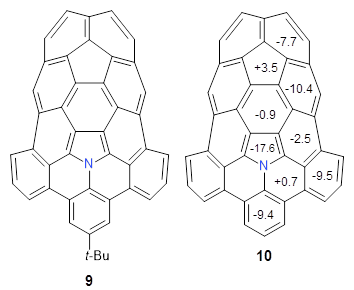
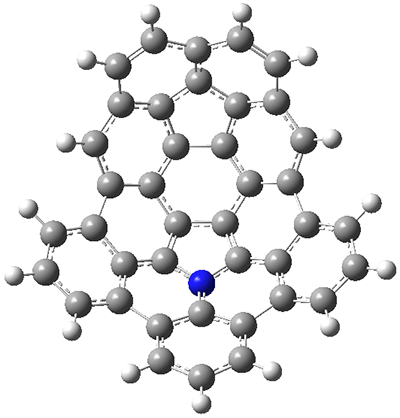
9.
5 The x-ray crystal structure shows a curved bowl shape with
Cs symmetry. NICS(0) values were computed for the parent molecule
10 B3LYP/6-31G(d). These values are shown in Scheme 1 and the geometry is shown in Figure 4. The 6-member rings that surround the azacylopentadienyl ring all have NICS(0) near zero, which suggests significant bond localisation.
Scheme 1. NICS(0) values of 10
Figure 4. B3LYP/6-31G(d) optimized structure of 10.
Our understanding of what aromaticity really means is constantly being challenged!
References
2. Ho, P. S.; Kit, C. C.; Jiye, L.; Zhifeng, L.; Qian, M., "A Dipleiadiene-Embedded Aromatic Saddle Consisting
of 86 Carbon Atoms."
Angew. Chem. Int. Ed. 2018,
57, 1581-1586, DOI:
10.1002/anie.201711437.
3. Yin, C. K.; Kit, C. C.; Zhifeng, L.; Qian, M., "A Twisted Nanographene Consisting of 96 Carbon Atoms."
Angew. Chem. Int. Ed. 2017,
56, 9003-9007, DOI:
10.1002/anie.201703754.
4. Koki, I.; Jennie, L.; Ryo, K.; Sota, S.; Hiroyuki, I., "Fluctuating Carbonaceous Networks with a Persistent
Molecular Shape: A Saddle-Shaped Geodesic Framework of 1,3,5-Trisubstituted Benzene (Phenine)."
Angew. Chem. Int. Ed. 2018,
57, 8555-8559, DOI:
10.1002/anie.201803984.
5. Yuki, T.; Shingo, I.; Kyoko, N., "A Hybrid of Corannulene and Azacorannulene: Synthesis of a Highly Curved Nitrogen-Containing Buckybowl."
Angew. Chem. Int. Ed. 2018,
57, 9818-9822, DOI:
10.1002/anie.201805678.
InChIs
1: InChI=1S/C134H128/c1-123(2,3)57-37-65-66-38-58(124(4,5)6)42-70-74-46-62(128(16,17)18)50-82-94(74)110-106(90(66)70)105-89(65)69(41-57)73-45-61(127(13,14)15)49-81-93(73)109(105)119-113-97(81)85(131(25,26)27)53-77-78-54-87(133(31,32)33)99-83-51-63(129(19,20)21)47-75-71-43-59(125(7,8)9)39-67-68-40-60(126(10,11)12)44-72-76-48-64(130(22,23)24)52-84-96(76)112-108(92(68)72)107(91(67)71)111(95(75)83)121-115(99)103(78)118-104-80(56-88(134(34,35)36)100(84)116(104)122(112)121)79-55-86(132(28,29)30)98(82)114(120(110)119)102(79)117(118)101(77)113/h37-56H,1-36H3
InChIKey=GKUTUWMASUJSFD-UHFFFAOYSA-N
2: InChI=1S/C86H32/c1-9-33-34-10-2-14-38-42-18-6-22-46-50-26-30-55-56-32-28-52-48-24-8-20-44-40-16-4-12-36-35-11-3-15-39-43-19-7-23-47-51-27-31-54-53-29-25-49-45-21-5-17-41-37(13-1)57(33)73-74(58(34)38)78(62(42)46)84-70(50)66(55)81(65(53)69(49)83(84)77(73)61(41)45)82-67(54)71(51)85-79(63(43)47)75(59(35)39)76(60(36)40)80(64(44)48)86(85)72(52)68(56)82/h1-32H
InChIKey=MXCDWJZMTKLBDM-UHFFFAOYSA-N
3: InChI=1S/C18H12/c1-2-6-14-11-12-16-8-4-3-7-15-10-9-13(5-1)17(14)18(15)16/h1-12H
InChIKey=KVJJNMIHWIRGRP-UHFFFAOYSA-N
4: InChI=1S/C132H108O4/c1-125(2,3)53-29-65-66-30-54(126(4,5)6)34-70-74-38-58(130(16,17)18)42-78-86-46-82-63-51-91(135-27)92(136-28)52-64(63)84-48-88-80-44-60(132(22,23)24)40-76-72-36-56(128(10,11)12)32-68-67-31-55(127(7,8)9)35-71-75-39-59(131(19,20)21)43-79-87-47-83-62-50-90(134-26)89(133-25)49-61(62)81-45-85-77-41-57(129(13,14)15)37-73-69(33-53)93(65)109-110(94(66)70)114(98(74)78)122-106(86)118-103(82)104(84)120-108(88)124-116(100(76)80)112(96(68)72)111(95(67)71)115(99(75)79)123(124)107(87)119(120)102(83)101(81)117(118)105(85)121(122)113(109)97(73)77/h29-52H,1-28H3
InChIKey=ZLPRACZKLACDHX-UHFFFAOYSA-N
5: InChI=1S/C108H60O4/c1-37-13-49-50-14-38(2)18-54-58-22-42(6)26-62-70-30-66-47-35-75(111-11)76(112-12)36-48(47)68-32-72-64-28-44(8)24-60-56-20-40(4)16-52-51-15-39(3)19-55-59-23-43(7)27-63-71-31-67-46-34-74(110-10)73(109-9)33-45(46)65-29-69-61-25-41(5)21-57-53(17-37)77(49)93-94(78(50)54)98(82(58)62)106-90(70)102-87(66)88(68)104-92(72)108-100(84(60)64)96(80(52)56)95(79(51)55)99(83(59)63)107(108)91(71)103(104)86(67)85(65)101(102)89(69)105(106)97(93)81(57)61/h13-36H,1-12H3
InChIKey=ZSIVUKSPPZUSQL-UHFFFAOYSA-N
6: InChI=1S/C32H16/c1-2-18-5-6-20-9-11-22-13-15-24-16-14-23-12-10-21-8-7-19-4-3-17(1)25-26(18)28(20)30(22)32(24)31(23)29(21)27(19)25/h1-16H
InChIkey=BASWMOIVIHXTRC-UHFFFAOYSA-N
7: InChI=1S/C224H210/c1-211(2,3)197-99-169-85-183(113-197)184-86-170(100-198(114-184)212(4,5)6)157-66-149-67-158(79-157)172-88-187(117-200(102-172)214(10,11)12)188-90-174(104-202(118-188)216(16,17)18)161-70-151-71-162(81-161)176-92-191(121-204(106-176)218(22,23)24)193-95-179(109-207(123-193)221(31,32)33)165-74-153-75-166(83-165)180-96-195(125-208(110-180)222(34,35)36)196-98-182(112-210(126-196)224(40,41)42)168-77-154-76-167(84-168)181-97-194(124-209(111-181)223(37,38)39)192-94-178(108-206(122-192)220(28,29)30)164-73-152-72-163(82-164)177-93-190(120-205(107-177)219(25,26)27)189-91-175(105-203(119-189)217(19,20)21)160-69-150-68-159(80-160)173-89-186(116-201(103-173)215(13,14)15)185-87-171(101-199(115-185)213(7,8)9)156-65-148(64-155(169)78-156)141-50-127-43-128(51-141)130-45-132(55-143(150)53-130)134-47-136(59-145(152)57-134)138-49-140(63-147(154)61-138)139-48-137(60-146(153)62-139)135-46-133(56-144(151)58-135)131-44-129(127)52-142(149)54-131/h43-126H,1-42H3
InChIKey=ZDDKJXIESSWTIA-UHFFFAOYSA-N
8: InChI=1S/C182H126/c1-99-15-113-43-127(29-99)141-57-142-65-155(64-141)162-78-169-92-170(79-162)172-82-164-83-174(94-172)176-85-166-87-178(96-176)180-89-168-91-182(98-180)181-90-167-88-179(97-181)177-86-165-84-175(95-177)173-81-163(80-171(169)93-173)156-66-143(128-30-100(2)16-114(113)44-128)58-144(67-156)130-32-103(5)19-117(47-130)118-20-104(6)34-132(48-118)147-60-148(71-158(165)70-147)134-36-107(9)23-121(51-134)123-25-109(11)39-137(53-123)151-62-152(75-160(167)74-151)138-40-111(13)27-125(55-138)126-28-112(14)42-140(56-126)154-63-153(76-161(168)77-154)139-41-110(12)26-124(54-139)122-24-108(10)38-136(52-122)150-61-149(72-159(166)73-150)135-37-106(8)22-120(50-135)119-21-105(7)35-133(49-119)146-59-145(68-157(164)69-146)131-33-102(4)18-116(46-131)115-17-101(3)31-129(142)45-115/h15-98H,1-14H3
InChIKey=FJHGGHOTCCNJNI-UHFFFAOYSA-N
9: InChI=1S/C44H23N/c1-44(2,3)21-16-28-24-8-4-6-22-26-14-19-12-10-18-11-13-20-15-27-23-7-5-9-25-29(17-21)41(28)45-42(33(22)24)39-35(26)37-31(19)30(18)32(20)38(37)36(27)40(39)43(45)34(23)25/h4-17H,1-3H3
InChIKey=QHBWEZKXFSKCSM-UHFFFAOYSA-N
10: InChI=1S/C40H15N/c1-4-19-23-8-3-9-24-20-5-2-7-22-26-15-18-13-11-16-10-12-17-14-25-21(6-1)30(19)39-36-32(25)34-28(17)27(16)29(18)35(34)33(26)37(36)40(31(20)22)41(39)38(23)24/h1-15H
InChIKey=XWSUADIIRLXSBY-UHFFFAOYSA-N

'
This work is licensed under a
Creative Commons Attribution-NoDerivs 3.0 Unported License.