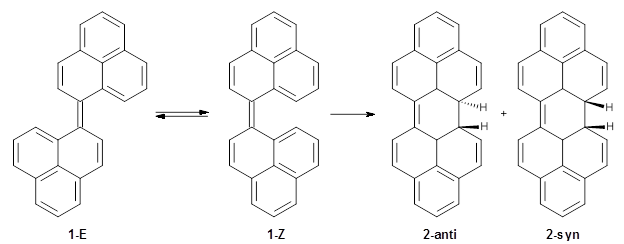
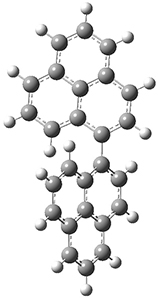
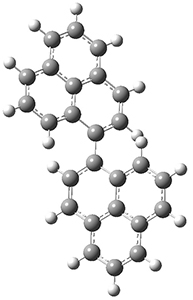
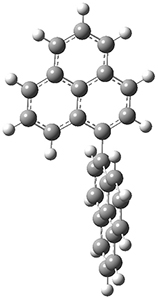
Yang, Y.-F.; Liang, Y.; Liu, F.; Houk, K. N. J. Am. Chem. Soc. 2016,138, 1660-1667
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
 '
'
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach
Reposted from Computational Organic Chemistry with permission
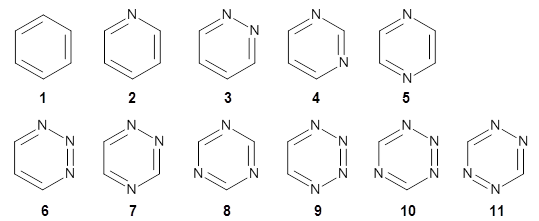
Houk has examined the Diels-Alder reaction involving ethene with benzene 1 and all of its aza-substituted isomers having four or fewer nitrogen atoms 2-11.1 The reactions were computed at M06-2X/6-311+G(d,p).
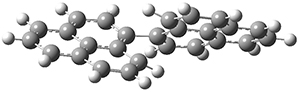
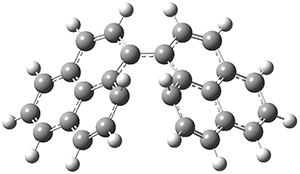
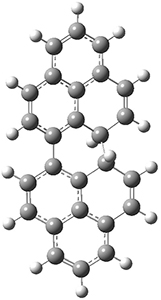
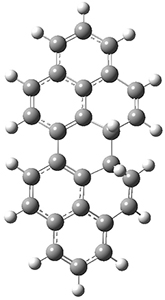
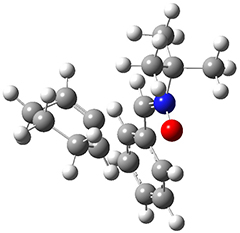
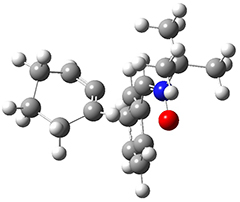
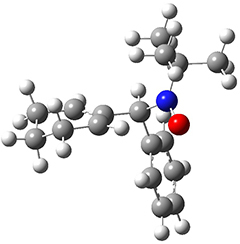
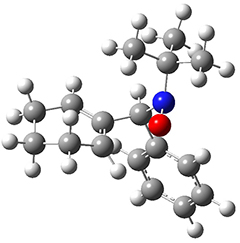
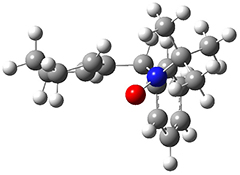
All of the possible Diels-Alder reactions were examined, and they can be classified in terms of whether two new C-C bonds are formed, one new C-C and one new C-N bond are formed, or two new C-N bonds are formed. Representative transition states of these three reaction types are shown in Figure 1, using the reaction of 7 with ethene.
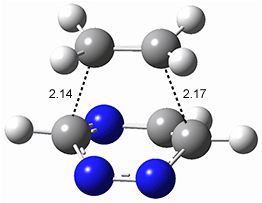 | 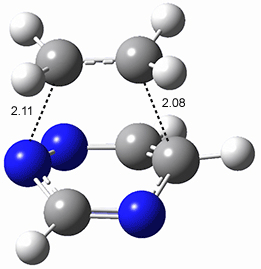 | 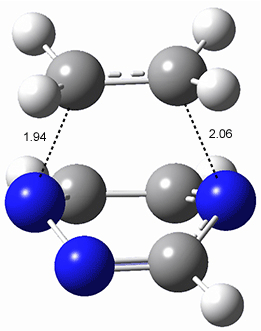 |
Figure 1. M06-2X/6-311+G(d,p) optimized transition states for the Diels-Alders reactions of 7 with ethene.
A number of interesting trends are revealed. For a given type of reaction (as defined above), as more nitrogens are introduced into the ring, the activation energy decreases. Forming two C-C bonds has a lower barrier than forming a C-C and a C-N, which has a lower barrier than forming two C-N bonds. The activation barriers are linearly related to the aromaticity of the ring defined by either NICS(0) or aromatic stabilization energy, with the barrier decreasing with decreasing aromaticity. The barrier is also linearly related to the exothermicity of the reaction.
The activation barrier is also linearly related to the distortion energy. With increasing nitrogen substitution, the ring becomes less aromatic, and therefore more readily distorted from planarity to adopt the transition state structure.
References
(1) Yang, Y.-F.; Liang, Y.; Liu, F.; Houk, K. N. "Diels–Alder Reactivities of Benzene, Pyridine, and Di-, Tri-, and Tetrazines: The Roles of Geometrical Distortions and Orbital Interactions," J. Am. Chem. Soc. 2016,138, 1660-1667, DOI: 10.1021/jacs.5b12054.
'This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.