Lee-Ping Wang, Alexey Titov, Robert McGibbon, Fang Liu, Vijay S. Pande and Todd J. Martínez
Nature Chemistry 6, 1044–1048 (2014)
Contributed by Alán Aspuru-Guzik
When is quantum chemistry going to take the primary discovery role, rather than an explanatory one? This one of the questions asked in the recent Nature Chemistry paper by the groups of Vijay Pande and Todd Martínez at Stanford.
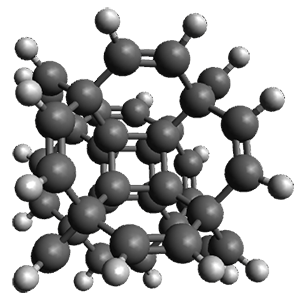
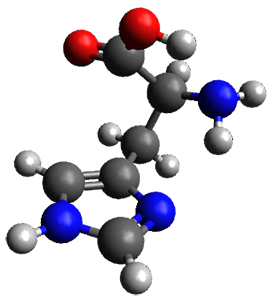
With the advent of fast GPGPU quantum chemistry in codes such as Terachem, it is practical to carry out medium-scale quantum dynamics simulations for a considerable amount of time. In this paper, the authors employ a spherical "piston" to compress and decompress high-temperature reaction mixtures, which in turn result in an accelerated process for triggering chemical reactions. Automated algorithms are employed to recognize the nature of the chemical transformations involved. The authors use this approach to identify novel mechanisms for the Urey-Miller origins of life experiment.
In this modern world of Facebook, Twitter, Youtube, Facebook and blogs, a video is worth more than a thousand words. The video below summarizes the paper very well.
This paper is indeed, one of my top 5 favorite papers in the field of computational chemistry of 2014.
 |
| Todd Martínez explaining the future of the nanoreactor in the QUITEL 2014 conference held in the Galápagos Islands in Ecuador. He suggested, in a joking fashion, that the nanoreactor could eventually evolve a flask with molecules into life ab initio. The virtual end product of the evolution seems to be the contributor of this post. |