James Shee, John L. Weber, David R. Reichman, Richard A. Friesner, and Shiwei Zhang (2022)
Highlighted by Jan Jensen

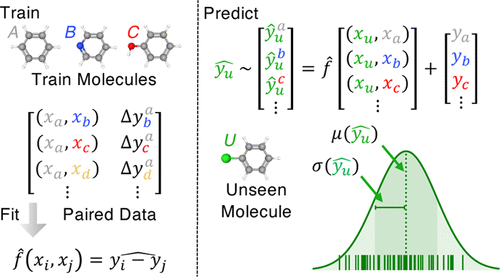
Figure 1 from this paper. (c) the authors
This paper highlights a big problem in the field of quantum chemistry and posits that a solution may be right around the corner. The problem is that we still can't routinely predict the thermochemistry of TM-containing compounds with the same degree of accuracy as we can for organic molecules. The main reason is that the former systems often have a high-degree of non-dynamic correlation which means that our CCSD(T) often does not give reliable results. We can model the non-dynamic correlation with CASSCF, but there is no good way to compute the dynamic correlation based on a CASSCF wavefunction. So when different DFT functional results give wildly different predictions for your TM-compound there is no way to tell which method, if any, if the best.
This paper argues that phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC) may be the solution to this problem. ph-AFQMC represents the ground state as a stochastic linear combination of Slater determinants mapped as open-ended random walks starting from a trial wavefunction. The method accounts for both non-dynamic and dynamic correlation and the paper argues that chemical accuracy can be achieved with a few hundred random walks, which can be run in parallel and on GPUs.
So what's missing? According to the authors some of the improvements needed include: more efficient ways of reaching the CBS limit, more efficient random walks and a general, automatable protocol to generate optimal trial wave functions. Let's hope these improvements will be made soon, so we can explore a much larger portion of chemical space with confidence.

This work is licensed under a Creative Commons Attribution 4.0 International License.