Marwin H. S. Segler, Mike Preuss, Mark P. Waller (2018)
Highlighted by Jan Jensen

The paper uses a Monte Carlo tree search (MCTS) algorithm (also used in AlphaGo Zero) to suggest retrosynthetic routes that were just as good as those proposed by expert organic chemist. Remarkably the underlying "expert knowledge" is automatically extracted from reaction databases into three neural networks. Thus, the method is referred to as 3N-MCTS.
At the core of this approach are two neural networks that can predict the probability of a molecule undergoing one of either 301,671 or 17,134 chemical transformations, the latter being more computationally efficient than the former. The networks were trained on tranformation rules from 12.4 million single-step reactions from the Reaxys chemistry database, i.e. determined automatically without human intervention.
I couldn't find any information about code availability.
Highlighted by Jan Jensen

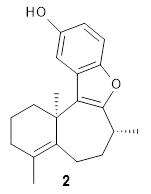
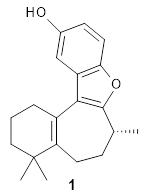
Figure 1 from the paper. Copyright 2018 Springer Nature
The paper uses a Monte Carlo tree search (MCTS) algorithm (also used in AlphaGo Zero) to suggest retrosynthetic routes that were just as good as those proposed by expert organic chemist. Remarkably the underlying "expert knowledge" is automatically extracted from reaction databases into three neural networks. Thus, the method is referred to as 3N-MCTS.
At the core of this approach are two neural networks that can predict the probability of a molecule undergoing one of either 301,671 or 17,134 chemical transformations, the latter being more computationally efficient than the former. The networks were trained on tranformation rules from 12.4 million single-step reactions from the Reaxys chemistry database, i.e. determined automatically without human intervention.
The retrosynthetic "game" is won if the target molecule can be completely decomposed into predefined precursor molecules within 25 retrosynthetic steps, where the 50 most probable chemical transformations are considered for each step. It is not practically possible to test all $50^{25} \approx 10^{40}$ possible retrosynthetic paths so a MCTS is used to search for the best path.
A MCTS starts by evaluating a number of paths randomly and then assigning likelihood scores to the early parts of the paths depending on whether the paths lead to winners or not. The process is then repeated except that the early steps in the path are chosen based on likelihood scores, which are continuously updated and added to unscored steps. The changing likelihood scores means that the search for new paths is directed towards the more promising areas of the path tree. I have given a short illustration of the process here. The process is repeated for a given number of steps and the path with the best set of likelihood scores is selected.
One of the tests of the method was a double blind study where experienced synthetic chemists were asked to choose between retrosynthetic routes developed by experts and by 3N-MCTS. The study found no clear preference!
A MCTS starts by evaluating a number of paths randomly and then assigning likelihood scores to the early parts of the paths depending on whether the paths lead to winners or not. The process is then repeated except that the early steps in the path are chosen based on likelihood scores, which are continuously updated and added to unscored steps. The changing likelihood scores means that the search for new paths is directed towards the more promising areas of the path tree. I have given a short illustration of the process here. The process is repeated for a given number of steps and the path with the best set of likelihood scores is selected.
One of the tests of the method was a double blind study where experienced synthetic chemists were asked to choose between retrosynthetic routes developed by experts and by 3N-MCTS. The study found no clear preference!
I couldn't find any information about code availability.