Julien Steffen and Bernd Hartke 2017
Highlighted by Jan Jensen

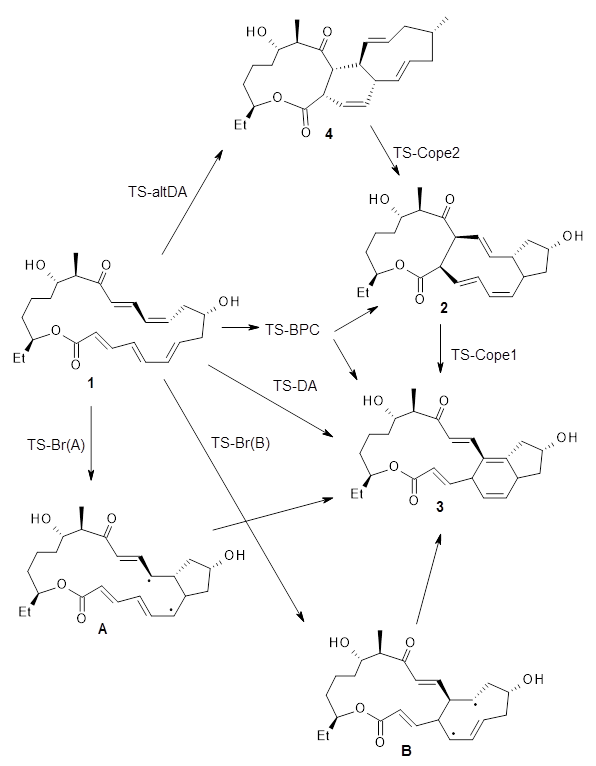
A few years ago I highlighted Grimme's General Quantum Mechanically Derived Force Field (QMDFF) - a black box approach that gives you a system-specific force field from a single QM Hessian calculation. I missed the fact that Hartke and Grimme extended this approach to TSs using EVB, a year later. This EVB-QMDFF approach constructs EVB potentials connecting each pair of minima described by QMDFF. To get the EVB parameters you need to supply the TS and 10-100 energies (and possibly 5-10 Hessian calculations) along the reaction path, depending on how complex an EVB potential is needed to describe the reaction.
What's the point of a system-specific reactive force field when you already have the TS and reaction path? Well, Steffen and Hartke show is that EVB-QMDFF can be used to perform the additional calculations needed for, for example, variational TS theory or ring polymer MD calculations to get more accurate rate constants.
Furthermore, just like for QMDFF for minima you could do all this for one conformation of ligands and use EVB-QMDFF for a conformer search or use the gas phase parameterized model to study the effect of explicit solvation. It might even be possible to parameterize EVB-QMDFF using small ligands and then model the effect of larger ligands using the QMDFF parameters obtained for the minima. However, all these potential uses still need to be tested.
I thank Jean-Philip Piquemal for bringing this paper to my attention

This work is licensed under a Creative Commons Attribution 4.0 International License.
Highlighted by Jan Jensen

Figure 1 from the paper. A flowchart of the EVB-QMDFF program implemented in this work, for the case of a DG-EVB-QMDFF calculation.
A few years ago I highlighted Grimme's General Quantum Mechanically Derived Force Field (QMDFF) - a black box approach that gives you a system-specific force field from a single QM Hessian calculation. I missed the fact that Hartke and Grimme extended this approach to TSs using EVB, a year later. This EVB-QMDFF approach constructs EVB potentials connecting each pair of minima described by QMDFF. To get the EVB parameters you need to supply the TS and 10-100 energies (and possibly 5-10 Hessian calculations) along the reaction path, depending on how complex an EVB potential is needed to describe the reaction.
What's the point of a system-specific reactive force field when you already have the TS and reaction path? Well, Steffen and Hartke show is that EVB-QMDFF can be used to perform the additional calculations needed for, for example, variational TS theory or ring polymer MD calculations to get more accurate rate constants.
Furthermore, just like for QMDFF for minima you could do all this for one conformation of ligands and use EVB-QMDFF for a conformer search or use the gas phase parameterized model to study the effect of explicit solvation. It might even be possible to parameterize EVB-QMDFF using small ligands and then model the effect of larger ligands using the QMDFF parameters obtained for the minima. However, all these potential uses still need to be tested.
I thank Jean-Philip Piquemal for bringing this paper to my attention
This work is licensed under a Creative Commons Attribution 4.0 International License.