Hotta, K.; Chen, X.; Paton, R. S.; Minami, A.; Li, H.; Swaminathan, K.; Mathews, I. I.; Watanabe, K.; Oikawa, H.; Houk, K. N.; Kim, C.-Y. Nature, 2012, 483, 355 (Paywall)
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
Biosynthesis of ladder polyethers is the topic of a very nice experimental/computational study by Chen and Houk.1 The x-ray structure of the enzyme that catalyzes the nucleophilic attack on epoxides to create the 6-member ring ether was determined, but the geometry did not completely indicate the mechanism.
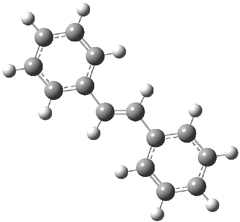
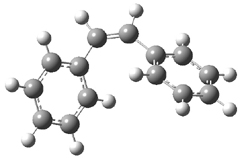
Gas phase computations of the 5-exo-tet and 6-endo-tet ring openings of 1 were examined for both the acid and base catalyzed routes at B2PLYP/6-311++G(d,p)//B2PLYP/6-31G(d).
The results are summarized in Figure 1. Basically, as expected by Baldwin’s rules, the closure to the tetrahydrofuran (5-exo-tet) is favored under both catalyzed conditions. However, the preference is small under base conditions, with the difference in the free energy of activation of only 1.2 kcal mol-1.

Figure 1. Gas phase energies (kcal mol-1) for the acid an base catalyzed reactions of 1 to 2 or 3.
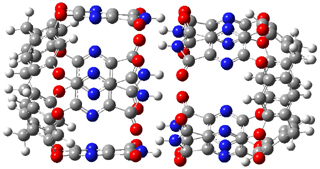
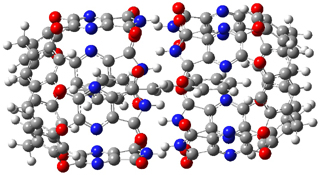
The enzyme Lsd19B produces just the analogue of 3. So, the two regioisomeric TSs were reoptimized with an aspartic acid group and a tyrosine group in the positions they occupy in the active site of the enzyme Lsd19b. The two resulting transition states, evaluated at B2LYP/6-311++G(d,p)//MO6-2x/6-31G(d), are shown in Figure 2. The activation energy for the 6-endo-tet reaction is 18.0 kcal mol-1, 2.5 kcal mol-1 lower than for the 5-exo-tet route. This energy difference would give rise to a 100:1 selectivity for the tetrahydropyran product, in accord with experiment. The enzyme preorganizes for and favors the base catalyzed path that leads to 3.
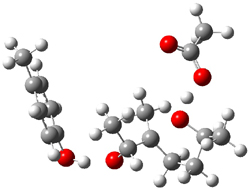 ΔG‡ = 20.5 |
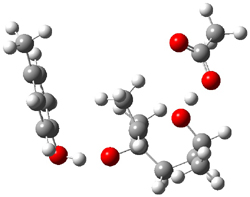 ΔG‡ = 18.0 |
Figure 2. Transition state models of the active site. Activation energies in kcal mol-1.
References
(1) Hotta, K.; Chen, X.; Paton, R. S.; Minami, A.; Li, H.; Swaminathan, K.; Mathews, I. I.; Watanabe, K.; Oikawa, H.; Houk, K. N.; Kim, C.-Y., "Enzymatic catalysis of anti-Baldwin ring closure in polyether biosynthesis," Nature, 2012, 483, 355-358, DOI: 10.1038/nature10865.
InChIs
1: InChI=1/C8H16O2/c1-6(9)4-5-8(3)7(2)10-8/h6-7,9H,4-5H2,1-3H3/t6-,7?,8+/m0/s1
InChIKey=YEAGKBPXLVGCPK-YPVSKDHRBB
2: InChI=1/C8H16O2/c1-6-4-5-8(3,10-6)7(2)9/h6-7,9H,4-5H2,1-3H3/t6-,7-,8-/m0/s1
InChIKey=RRDMOYCGUXNJSL-FXQIFTODBG
3: InChI=1/C8H16O2/c1-6-4-5-8(3,9)7(2)10-6/h6-7,9H,4-5H2,1-3H3/t6-,7-,8+/m0/s1
InChIKey=WWLWIBPPUNCAQU-BIIVOSGPBQ

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.