Gilmore, K.; Manoharan, M.; Wu, J. I. C.; Schleyer, P. v. R.; Alabugin, I. V. J. Am. Chem. Soc.2012, 134, 10584–10594 (Paywall)
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
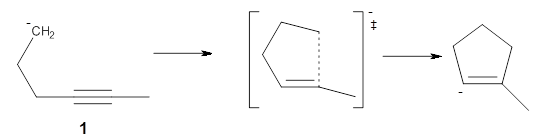
The activation energy for the 5-endo-dig reaction of the anion 1 is anomalously low compared to its 4-endo-dig and 6-endo-dig analogues. Furthermore, the TS is quite early, earlier than might be expected based on the Hammond Postulate. Alabugin and Schleyer have examined this reaction and found some interesting results.1
First, NICS(0) values for a series of related intermolecular anionic attack at alkynes show some interesting trends (Table 1). Two of the transition states look like they might be aromatic: the TSs for the 3-exo-dig and the 5-endo-dig reaction have NICS(0) values that are quite negative. However, given the geometry of these TSs, particularly the close proximity of the σ bonds to the ring center, one might be concerned about contamination of these orbitals. So, NICS(0)MOzz computations, which look at the tensor component perpendicular to the ring using just the π-MOs, shows that the 3-exo-dig is likely non-aromatic (NICS(0)MOzz is near zero), the TS for the 4-endo-dig reaction is antiaromatic (NICS(0)MOzz very positive) and the TS for the 5-endo-dig reaction is aromatic (NICS(0)MOzz is very negative. So this last reaction is the first example of an aromatic transition that is not for a pericyclic reaction!
Table 1. NICS(0) and NICS(0)MOzz for the TS of some anionic alkyne cyclizations.
NICS(0)
|
NICS(0)MOzz
| |
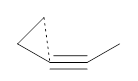 3-exo-dig |
-19.3
|
-1.6
|
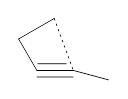 4-endo-dig |
1.8
|
23.9
|
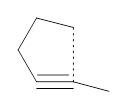 5-endo-dig (1) |
-15.2
|
-20.5
|
These authors argue that the reaction of 1 is an “aborted” sigmatropic shift. A normal pericyclic reaction is a single step with a single (concerted) transition state. An interrupted sigmatropic shift has an intermediate that lies higher in energy than the reactants, such as in the Bergman cyclization of an enediyne. The aborted sigmatropic shift has an intermediate that lies lower in energy than the reactants, such as in the cyclization of 1.
References
(1) Gilmore, K.; Manoharan, M.; Wu, J. I. C.; Schleyer, P. v. R.; Alabugin, I. V. "Aromatic Transition States in Nonpericyclic Reactions: Anionic 5-Endo Cyclizations Are Aborted Sigmatropic Shifts," J. Am. Chem. Soc.2012, 134, 10584–10594, DOI: 10.1021/ja303341b
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.