Bispericyclic reactions occur when two different pericyclic reactions merge to have a single transition state. An example of this is the joining of two [3,3]-sigmatopic rearrangements of
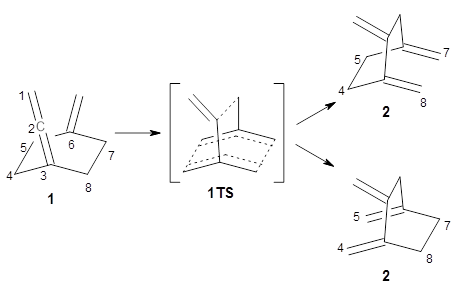
1 that merge to have a single transition state. Lopez, Faza, and Lopez have examined the dynamics of this reaction.
1
Because of the symmetry of the species along this reaction pathway, the products of the two different rearrangements are identical, and will be formed in equal amounts, though they are produced from a single transition state with the reaction pathway bifurcating due to a valley-ridge inflection post TS.
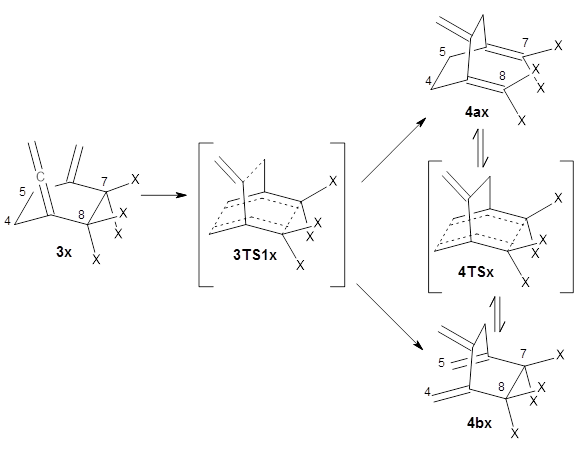
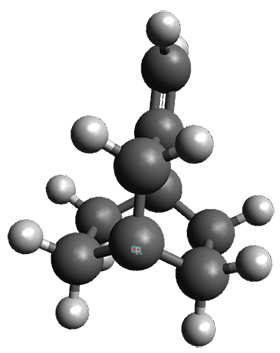
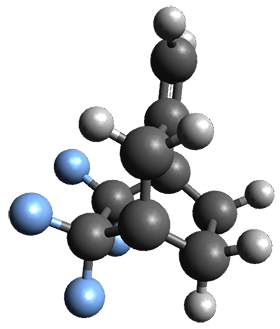
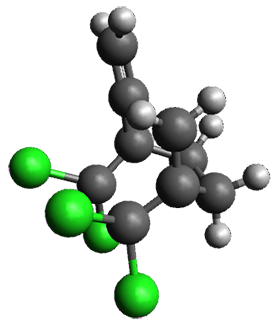
The interesting twist that is explored here is when 1 is substituted in order to break the symmetry. The authors have examined 3x with either fluorine, chlorine, or bromine. The critical points on the reactions surface were optimized at M06-2X/Def2TZVPP. In all three cases a single bispericyclic transition state 3TS1x is found, which leads to products 4a and 4b. A second transition state 4TSx corresponds to the [3,3]-rearrangement that interconverts the two products. The structures of 1TS, 3TS1F, and 3TS1Cl are shown in Figure 1.
Figure 1. M06-2X/Def2TZVPP optimized geometries of 1TS, 3TS1F, and 3TS1Cl.
The halogen substitution breaks the symmetry of the reaction path. This leads to a number of important changes. First, the C4-C5 and C7-C8 distances, which are identical in 1TS, are different in the halogen cases. Interestingly, the distortions are dependent on the halogen: in 3TS1F C4-C5 is 0.2 Å longer than C7-C8, but in 3TS1Cl C7-C8 is much longer (by 0.65 Å) than C4-C5. Second, the products are no longer equivalent with the halogen substitution. Again, this is halogen dependent: 4bF is 4.0 kcal mol-1 lower in energy than 4aF, while 4aCl is 8.2 kcal mol-1 lower than 4bCl.
These difference manifest in very different reaction dynamics. With trajectories initiated at the first (bispericyclic) transiting state, 89% end at
4bF and 9% end at
4aF, a ratio far from unity that might be expected from both products resulting from passage through the same TS. The situation is even more extreme for the chlorine case, where all 200 trajectories end in
4aCl. This is yet another example of the role that dynamics play in reaction outcomes (see these many
previous posts).
References
1) Villar López, R.; Faza, O. N.; Silva López, C., "Dynamic Effects Responsible for High Selectivity in a [3,3] Sigmatropic Rearrangement Featuring a Bispericyclic Transition State."
J. Org. Chem. 2017, 82 (9), 4758-4765, DOI:
10.1021/acs.joc.7b00425.
InChIs
1: InChI=1S/C9H12/c1-3-9-6-4-8(2)5-7-9/h1-2,4-7H2
InChIKey=RRXCPJIEZVQPSZ-UHFFFAOYSA-N
2: InChI<=1S/C9H12/c1-7-4-5-8(2)9(3)6-7/h1-6H2
InChIKey=AMBNQWVPTPHADI-UHFFFAOYSA-N
3F: InChI=1S/C9H8F4/c1-3-7-5-4-6(2)8(10,11)9(7,12)13/h1-2,4-5H2
InChIKey=VZFAQFJKHDWJDN-UHFFFAOYSA-N
3Cl: InChI=1S/C9H8Cl4/c1-3-7-5-4-6(2)8(10,11)9(7,12)13/h1-2,4-5H2
InChIKey=AIVUHFMHIMNOJB-UHFFFAOYSA-N
4aF: InChI=1S/C9H8F4/c1-5-4-6(8(10)11)2-3-7(5)9(12)13/h1-4H2
InChIKey=NAUUHIHYMAOMIF-UHFFFAOYSA-N
4aCl: InChI=1S/C9H8Cl4/c1-5-4-6(8(10)11)2-3-7(5)9(12)13/h1-4H2
InChIKey=MMCKDJXQYSGQEH-UHFFFAOYSA-N
4bF: InChI=1S/C9H8F4/c1-5-4-6(2)8(10,11)9(12,13)7(5)3/h1-4H2
InChIKey=LMFNAIRCNARWSX-UHFFFAOYSA-N
4bCl: InChI=1S/C9H8Cl4/c1-5-4-6(2)8(10,11)9(12,13)7(5)3/h1-4H2
InChIKey=NOFFASDSCUGRTP-UHFFFAOYSA-N

'
This work is licensed under a
Creative Commons Attribution-NoDerivs 3.0 Unported License.