Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
Here is an interesting twist on using computations in conjunction with experimental NMR to solve for molecular structure. I have blogged a number of times on comparing computed chemical shifts with experimental values to identify structure, and also on using the comparison of computed and experimental coupling constants to accomplish this purpose.
Butts and Bifulco were interested in the structure of conicasterol F 1 and opted to make two sets of comparison.1 The first uses the traditional approach of comparing the computed and experimental 13C chemical shifts. The second comparison uses the distances between protons, coming from the optimized structure and the rotating-frame nuclear Overhauser effect (ROE).
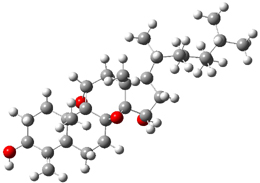
Standard analysis of the NMR spectra of 1 allowed for the determination of all of the stereochemistry except for the epoxy ring at C8 and C14. The possible options are shown as 1a and 1b. The optimized geometries (MPW1PW91/6-31G) of these two diastereomers are shown in Figure 1.
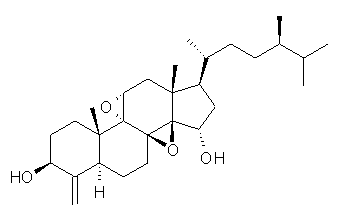 1a |
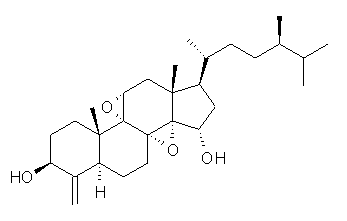 1b |
 |
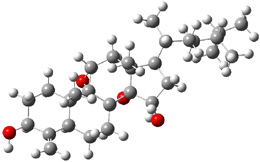 |
Figure 1. Optimized geometries of 1a and 1b.
References
(1) Chini, M. G.; Jones, C. R.; Zampella, A.; D’Auria, M. V.; Renga, B.; Fiorucci, S.; Butts, C. P.; Bifulco, G., "Quantitative NMR-Derived Interproton Distances Combined with Quantum Mechanical Calculations of 13C Chemical Shifts in the Stereochemical Determination of Conicasterol F, a Nuclear Receptor Ligand from Theonella swinhoei," J. Org. Chem., 2012, 77, 1489-1496, DOI: 10.1021/jo2023763.
InChIs
1b: InChI=1/C29H46O4/c1-16(2)17(3)8-9-18(4)21-14-23(31)28-26(21,7)15-24-29(32-24)25(6)12-11-22(30)19(5)20(25)10-13-27(28,29)33-28/h16-18,20-24,30-31H,5,8-15H2,1-4,6-7H3/t17-,18-,20+,21-,22+,23+,24-,25+,26-,27+,28+,29+/m1/s1
InChIKey=XTWHHLLDFQALAM-XAIGNWORBC

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment