Shokri, A.; Abedin, A.; Fattahi, A.; Kass, S. R. J. Am. Chem. Soc. 2012, 134, 10646 (Paywall)
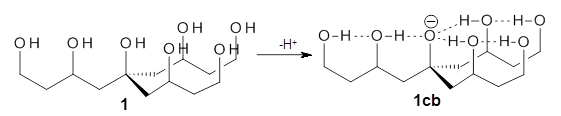
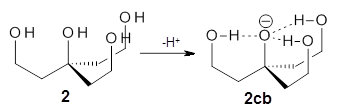
Can a hydrogen bonding network affect acidity? Kass has examined the polyol 1 whose conjugate base1cb can potentially be stabilized by a large hydrogen bonding network.1 Kass had previously found a significant acidy enhancement in comparing t-butanol (ΔG(deprotonation) = 369.2 kcal mol-1) with that of 2 (ΔG(deprotonation) = 334.4 kcal mol-1).2
Table 1 lists the computed and experimental free energies of deprotonation of 1. The experimental values are computed at M06-2x/maug-cc-pVT(+d)Z. The structure of 1cb is drawn in Figure 1.
Table 1. Computed and experimental free energies (kcal mol-1 of deprotonation of some alcohols.
MO6-2x
|
Expt
| |
t-butanol
|
368.6
|
369.3
|
2
|
335.0
|
334.4
|
1
|
320.2
|
313.5
|
The difference in the acidity of t-butanol and 2, some 30 kcal mol-1, reflects the stability afforded by three intramolecular hydrogen bonds to the oxyanion. In going from 2cb to 1cb, each of the hydroxyl groups that donate to the oxyanion act as the acceptor of a hydrogen bond from the more removed hydroxyl groups. There is in effect a first and second layer of hydrogen bond network in 1cb. These secondary hydrogen bonds lead to further stabilization of the anion, as reflected in the diminished DPE of 1 over 2: 320.2 vs. 335.0 kcal mol-1. Note that this secondary layer does not stabilize the anion to the same degree as the primary layer, but nonetheless its effect is large and quite striking.
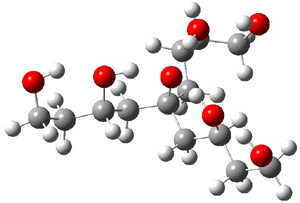
1cb
|
Figure 1. M06-2x/maug-cc-pVT(+d)Z optimized structure of 1cb.
Even in solution these more remote hydrogen bonds can stabilize the anion. So, using the CPCM approach and modeling DMSO, 2 is predicted have a pKa that is 15 units below that of t-butanol, and 1is predicted to be 3 pKa units more acidic than 2. Experiments verify this prediction with the pKas of 16.1 for 2 and 11.4 for 1.
References
(1) Shokri, A.; Abedin, A.; Fattahi, A.; Kass, S. R. "Effect of Hydrogen Bonds on pKa Values: Importance of Networking," J. Am. Chem. Soc. 2012, 134, 10646-10650, DOI: 10.1021/ja3037349
(2) Tian, Z.; Fattahi, A.; Lis, L.; Kass, S. R. "Single-Centered Hydrogen-Bonded Enhanced Acidity (SHEA) Acids: A New Class of Brønsted Acids," J. Am. Chem. Soc. 2009, 131, 16984-16988, DOI:10.1021/ja9075106
InChIs
1: InChI=1S/C13H28O7/c14-4-1-10(17)7-13(20,8-11(18)2-5-15)9-12(19)3-6-16/h10-12,14-20H,1-9H2
InChIKey=HGTVPOTWAYDRSM-UHFFFAOYSA-N
InChIKey=HGTVPOTWAYDRSM-UHFFFAOYSA-N
1cb: InChI=1S/C13H27O7/c14-4-1-10(17)7-13(20,8-11(18)2-5-15)9-12(19)3-6-16/h10-12,14-19H,1-9H2/q-1
InChIKey=UQPPTNIHRICITD-UHFFFAOYSA-N
InChIKey=UQPPTNIHRICITD-UHFFFAOYSA-N
2: InChI=1S/C7H16O4/c8-4-1-7(11,2-5-9)3-6-10/h8-11H,1-6H2
InChIKey=FAQWYKIIWYVDPQ-UHFFFAOYSA-N
InChIKey=FAQWYKIIWYVDPQ-UHFFFAOYSA-N
2cb: InChI=1S/C7H15O4/c8-4-1-7(11,2-5-9)3-6-10/h8-10H,1-6H2/q-1
InChIKey=WSCPTRIAWKZJFZ-UHFFFAOYSA-N
InChIKey=WSCPTRIAWKZJFZ-UHFFFAOYSA-N

