Kawasumi, K.; Zhang, Q.; Segawa, Y.; Scott, L. T.; Itami, K. Nat Chem 2013, advance online publication
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission

This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bachrach.
Reposted from Computational Organic Chemistry with permission
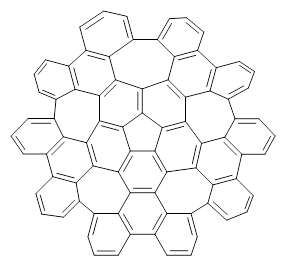
Scott and Itami report on a graphene fragment that is highly warped.1 They have prepared 1 by three separate procedures, one of which starts with corranulene and in two steps makes the product!
 1 |
The five 7-member rings warp the structure so that it is non-planar. In fact the molecule has negative curvature, reminiscent of a riding saddle. They report the x-ray structure, outside of the fullerenes, the largest hydrocarbon reported by x-ray crystallography. Because of its non-planar geometry, 1 does not pack well and so it is soluble in a variety of solvents.
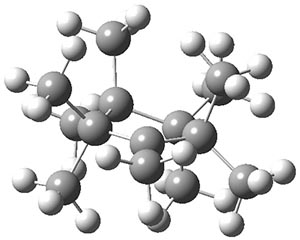
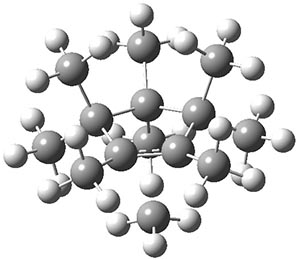
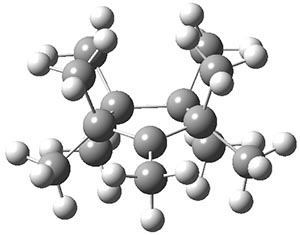
The authors have obtained the structure of 1 at B3LYP/6-31G(d), shown in Figure 1. The central corranulene component is a shallow bowl, much less shallow than in corranulene itself. This suggests that the compound might flip with a relatively low barrier. The computed barrier is only 1.7 kcal mol-1. Due to the negative curvatures associated with the seven-member rings, 1 is chiral and the ring flipping process leaves the chirality unchanged. A second transition was located that leads to racemization through a transition state of Cs symmetry. The barrier for this racemization is computed to be 18.9 kcal mol-1. Variable temperature 1H NMR analysis does show that at room temperature 1 (substituted with one t-butyl ring on each of the ten exterior phenyl rings) undergoes rapid motion that equilibrates all of the protons. However, at lower temperature the signals for ring protons do separate. This leads to the barrier or the racemization process of 13.6 kcal mol-1. The ring flip is not frozen out at the temperatures explored.
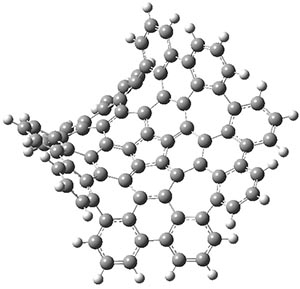
1
|
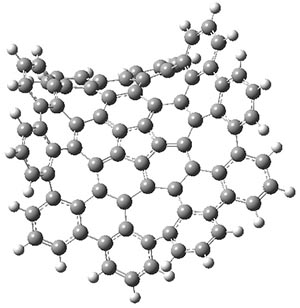
1-TSflip
|
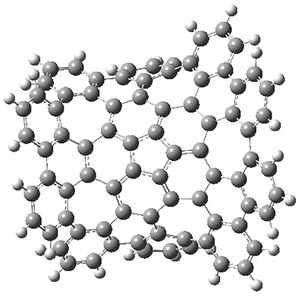
1-TSrac
|
Figure 1. B3LYP/6-31G(d) optimized structures of 1 and the transition states for flipping and racemization. (Remember that all structures in my blog are active – click on them to run Jmol and manipulate the 3-D structure.)
Compound 1 is an example of a very interesting negative curvature hydrocarbon, especially unusual for what might be considered an aromatic compound.
References
(1) Kawasumi, K.; Zhang, Q.; Segawa, Y.; Scott, L. T.; Itami, K. "A grossly warped nanographene and the consequences of multiple odd-membered-ring defects," Nat Chem 2013, advance online publication, DOI: 10.1038/nchem.1704.
InChIs
1: InChI=1S/C80H30/c1-11-31-32-12-2-23-43-44-24-5-15-35-36-16-6-27-47-48-28-9-19-39-40-20-10-30-50-49-29-8-18-38-37-17-7-26-46-45-25-4-14-34-33-13-3-22-42-41(21-1)51(31)61-62(52(32)43)72-65(55(35)44)66(56(36)47)74-69(59(39)48)70(60(40)50)75-68(58(38)49)67(57(37)46)73-64(54(34)45)63(53(33)42)71(61)76-77(72)79(74)80(75)78(73)76/h1-30H
InChIKey=KMAOOAQTQHCOHV-UHFFFAOYSA-N
InChIKey=KMAOOAQTQHCOHV-UHFFFAOYSA-N
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.