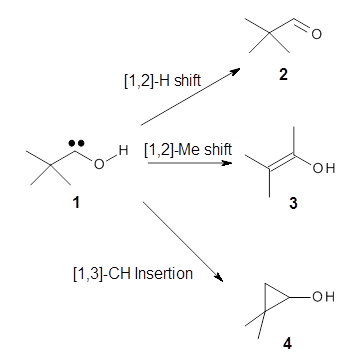
Nusret D. Yilmazer and Martin Korth Journal of Physical Chemistry B 2013, 117, 8075
Contributed by +Jan Jensen
Contributed by +Jan Jensen
This paper provides an extremely thorough comparison of electronic protein-ligand interaction energies computed using MM force fields, semi-empirical methods, and DFT.
About 700 protein-ligand complexes were selected from the PDBbind2007 database and four model systems of increasing size (including atoms up to 10 Å from a ligand atom) are created for each. DFT calculations were only possible for around 500 complexes using a 5 Å cutoff, and MM calculations were only possible for 352 complexes due to parameterization problems.
Single point energies for the protein-ligand complex and separated protein and ligand are then computed and used to compute interaction energies, which were then compared amongst each other. For example, systems of different size and be compared to monitor convergence and different methods can be compared for a given cutoff.
Some of the more interesting comparisons to me are PM6-DH+ vs BP86-D2/TZVP (R = 1.00) and MMFF94 vs BP86-D2/TZVP (R = 0.93). (Interestingly, the former value decreases to 0.92 if COSMO is included - that is unexpected to me.) While these R values are quite good the corresponding MAD values are quite bad: 14 and 57 kcal/mol, respectively! I find the former quite surprising since the error in the PM6-DH+ hydrogen bond strengths computed for the underlying training set are on the order of 2 kcal/mol. It could be non-additivity or more interactions between charged groups in the present set.
Acknowledgement: thanks to +Jimmy Charnley Kromann for alerting me to this paper