Xin, D.; Sader, C. A.; Chaudhary, O.; Jones, P.-J.; Wagner, K.; Tautermann, C. S.; Yang, Z.; Busacca, C. A.; Saraceno, R. A.; Fandrick, K. R.; Gonnella, N. C.; Horspool, K.; Hansen, G.; Senanayake, C. H., J. Org. Chem. 2017, ASAP
Contributed by Steven Bacharach
Reposted from Computational Organic Chemistry with permission
 '
'
This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
Contributed by Steven Bacharach
Reposted from Computational Organic Chemistry with permission
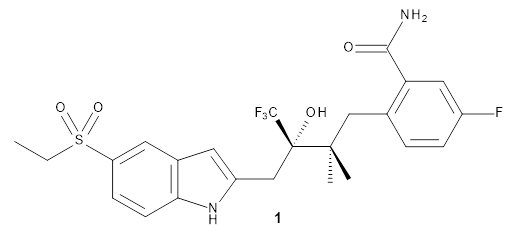
Here’s another take on automating a procedure for using computer 13C chemical shifts to assess chemical structure.1 (Have a look at these previous posts for some alternative methods and applications.) The approach here is to benchmark a few computational methods against a conformationally flexible drug-like molecule, in this case 1. A variety of conformations were optimized using the different computational methods, and 13C chemical shifts evaluated from a Boltzmann-weighted distribution. While the best agreement with the experimental chemical shifts (based on the root-mean-squared deviation) is with ωB97XD/cc-pVDZ, the authors opt for B3LYP/cc-pVDZ for its computational efficiency with only slightly poorer performance. (It should be note that WC04/cc-pVDZ, a functional designed for computing 13chemical shifts,2 is almost as good as ωB97XD/cc-pVDZ. Also, not mentioned in the article is the dramatically poorer performance of the pcS-2 basis set, despite the fact that it was parametrized3 for NMR computation!)
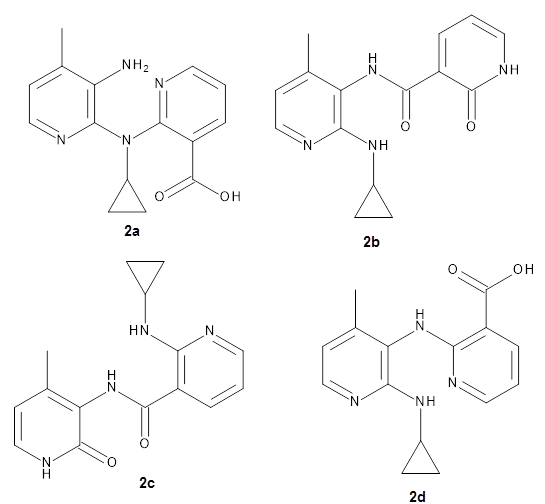
They apply the procedure to a number of test cases. For example, the HIV-1 reverse transcriptase inhibitor nevirapine hydrolyzes to a compound whose structure has been difficult to identify. The four proposed structures 2a-d were subjected to the computational method, and the 13C chemical shift RMSD for 2d is only 2.3ppm, significantly smaller than for the other 3 structures. Compound 2d was then synthesized and its NMR matches that of the nevirapine hydrolysis product.
References
1) Xin, D.; Sader, C. A.; Chaudhary, O.; Jones, P.-J.; Wagner, K.; Tautermann, C. S.; Yang, Z.; Busacca, C. A.; Saraceno, R. A.; Fandrick, K. R.; Gonnella, N. C.; Horspool, K.; Hansen, G.; Senanayake, C. H., "Development of a 13C NMR Chemical Shift Prediction Procedure Using B3LYP/cc-pVDZ and Empirically Derived Systematic Error Correction Terms: A Computational Small Molecule Structure Elucidation Method." J. Org. Chem. 2017, ASAP, DOI: 10.1021/acs.joc.7b00321.
2) Wiitala, K. W.; Hoye, T. R.; Cramer, C. J., “Hybrid Density Functional Methods Empirically Optimized for the Computation of 13C and 1H Chemical Shifts in Chloroform Solution,” J. Chem. Theory Comput. 2006, 2, 1085-1092, DOI: 10.1021/ct6001016.
3) Jensen, F., “Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods,” J. Chem. Theory Comput., 2008, 4, 719-727, DOI: 10.1021/ct800013z.
InChIs
1: InChI=1S/C24H26F4N2O4S/c1-4-35(33,34)18-7-8-20-15(10-18)9-17(30-20)13-23(32,24(26,27)28)22(2,3)12-14-5-6-16(25)11-19(14)21(29)31/h5-11,30,32H,4,12-13H2,1-3H3,(H2,29,31)/t23-/m0/s1
InChIKey=ILKZCEOVIFOUBJ-QHCPKHFHSA-N
InChIKey=ILKZCEOVIFOUBJ-QHCPKHFHSA-N
2d: InChI=1S/C15H16N4O2/c1-9-6-8-17-14(18-10-4-5-10)12(9)19-13-11(15(20)21)3-2-7-16-13/h2-3,6-8,10H,4-5H2,1H3,(H,16,19)(H,17,18)(H,20,21)
InChIKey=ZLFOGBWAZNUXAD-UHFFFAOYSA-N
InChIKey=ZLFOGBWAZNUXAD-UHFFFAOYSA-N
'This work is licensed under a Creative Commons Attribution-NoDerivs 3.0 Unported License.
No comments:
Post a Comment