Mackey, J. L.; Yang, Z.; Houk, K. N., "Dynamically concerted and stepwise trajectories of the Cope rearrangement of 1,5-hexadiene." Chem. Phys. Lett. 2017, 683, 253-257Yang, Z.; Zou, L.; Yu, Y.; Liu, F.; Dong, X.; Houk, K. N., "Molecular dynamics of the two-stage mechanism of cyclopentadiene dimerization: concerted or stepwise?" Chem. Phys. 2018, in pressYang, Z.; Dong, X.; Yu, Y.; Yu, P.; Li, Y.; Jamieson, C.; Houk, K. N., "Relationships between Product Ratios in Ambimodal Pericyclic Reactions and Bond Lengths in Transition Structures." J. Am. Chem. Soc. 2018, 140, 3061-3067Contributed by
Steven BacharachReposted from
Computational Organic Chemistry with permission
At the recent ACS meeting in New Orleans, Ken Houk spoke at the Dreyfus award session in honor of Michele Parrinello. Ken’s talk included discussion of some recent molecular dynamics studies of pericyclic reactions. Because of their similarities in approaches and observations, I will discuss three recent papers from his group (which Ken discussed in New Orleans) in this post.
The Cope rearrangement, a fundamental organic reaction, has been studied extensively by computational means (see Chapter 4.2 of my book). Mackey, Yang, and Houk examine the degenerate Cope rearrangement of 1,5-hexadiene with molecular dynamics at the (U)B3LYP/6-31G(d) level.
1 They examined 230 trajectories, and find that of the 95% of them that are reactive, 94% are trajectories that directly cross through the transition zone. By this, Houk means that the time gap between the breaking and forming C-C bonds is less than 60 fs, the time for one C-C bond vibration. The average time in the transition zone is 35 fs. This can be thought of as “dynamically concerted”. For the other few trajectories, a transient diradical with lifetime of about 100 fs is found.
The dimerization of cyclopentadiene finds the two [4+2] pathways merging into a single bispericylic transition state.
2 Only a small minority (13%) of the trajectories sample the region about the Cope rearrangement that interconverts the two mirror image dimers. These trajectories average about 60 fs in this space, which comes from the time separation between the formation of the two new C-C bonds. The majority of the trajectories quickly pass through the dimerization transition zone in about 18 fs, and avoid the Cope TS region entirely. These paths can be thought of as “dynamically concerted”, while the other set of trajectories are “dynamically stepwise”. It should be noted however that the value of
S2 in the Cope transition zone are zero and so no radicals are being formed.
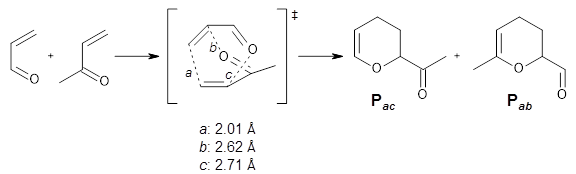
Finally, Yang, Dong, Yu, Yu, Li, Jamieson, and Houk examined 15 different reactions that involve ambimodal (i.e. bispericyclic) transition states.
3 They find a strong correlation between the differences in the bond lengths of the two possible new bond
vs. their product distribution. So for example, in the reaction shown in Scheme 1, bond
a is the one farthest along to forming. Bond
b is slightly shorter than bond
c. Which of these two is formed next is dependent on the dynamics, and it turns out the
Pab is formed from 73% of the trajectories while
Pac is formed only 23% of the time. This trend is seen across the 15 reaction, namely the shorter of bond
b or
c in the transition state leads to the larger product formation. When competing reactions involve bonds with differing elements, then a correlation can be found with bond order instead of with bond length.
Scheme 1
References
1) Mackey, J. L.; Yang, Z.; Houk, K. N., "Dynamically concerted and stepwise trajectories of the Cope rearrangement of 1,5-hexadiene."
Chem. Phys. Lett. 2017, 683, 253-257, DOI:
10.1016/j.cplett.2017.03.011.
2) Yang, Z.; Zou, L.; Yu, Y.; Liu, F.; Dong, X.; Houk, K. N., "Molecular dynamics of the two-stage mechanism of cyclopentadiene dimerization: concerted or stepwise?"
Chem. Phys. 2018, in press, DOI:
10.1016/j.chemphys.2018.02.020.
3) Yang, Z.; Dong, X.; Yu, Y.; Yu, P.; Li, Y.; Jamieson, C.; Houk, K. N., "Relationships between Product Ratios in Ambimodal Pericyclic Reactions and Bond Lengths in Transition Structures."
J. Am. Chem. Soc. 2018,140, 3061-3067, DOI:
10.1021/jacs.7b13562.

'
This work is licensed under a
Creative Commons Attribution-NoDerivs 3.0 Unported License.