Eugene E. Kwan and Richard Y. Liu (2015) DOI 10.1021/acs.jctc.5b00856

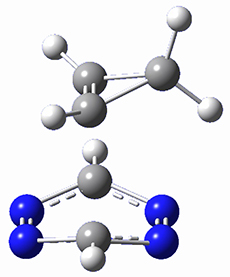
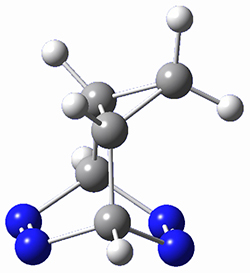
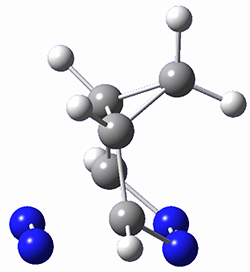
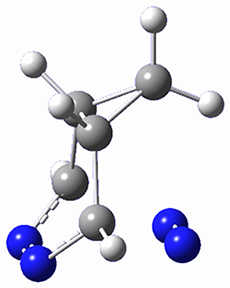
This paper presents a novel way of computing vibrational effects on chemical shifts. Existing methods generally compute such effects by displacing the coordinates along the normal modes to create PESs, computing the vibrational wavefunctions, computing chemical shifts for the displaced coordinates, and computing the chemical shifts as expectation values. However, it can be challenging to use this approach on low frequency modes since they require relatively large displacements which tend to distort the molecule in unphysical ways. This problem can, in part, be solved by using internal coordinates but results can wary greatly depending on which internal coordinates one chooses.
Kwan and Liu propose instead to perform a short (125 fs) B3LYP/MIDI! quasi-classical MD simulation "along" each normal mode and then compute an average chemical shift based on the trajectory. The MD simulation is "quasi-classical" in the sense that it is initialized based in the vibrational harmonic oscillator energy levels. The energy level is randomly selected from a Boltzmann distribution and 25 trajectories were found to be sufficient. Since the forces are computed at each point and used to determine the next displacement point unphysical distortions of the molecule are avoided. Another difference is that the chemical shifts are computed using a higher level of theory (B3LYP/cc-pVDZ) than that used to construct the PES.
The various choices for method, basis set, simulation length, number of trajectories, etc is tested extensively in the supplementary materials, which also contains a more thorough description of steps in the algorithm (page 96). Here the authors also state that they "are in the process of developing a user-friendly package for carrying out these calculations, which will be reported in due course."
Compared to the previously published methods for computing vibrational corrections this method is significantly more expensive in terms of energy and gradient evaluations. However, I wonder if the methods can be combined such that this method is used only for lower frequency modes that can problematic for the "displacement" methods. I also wonder if the method can be adapted to compute the anharmonic corrections to the enthalpy and entropy, which also can prove challenging for displacement-based methods.
The various choices for method, basis set, simulation length, number of trajectories, etc is tested extensively in the supplementary materials, which also contains a more thorough description of steps in the algorithm (page 96). Here the authors also state that they "are in the process of developing a user-friendly package for carrying out these calculations, which will be reported in due course."
Compared to the previously published methods for computing vibrational corrections this method is significantly more expensive in terms of energy and gradient evaluations. However, I wonder if the methods can be combined such that this method is used only for lower frequency modes that can problematic for the "displacement" methods. I also wonder if the method can be adapted to compute the anharmonic corrections to the enthalpy and entropy, which also can prove challenging for displacement-based methods.